-
Enantioselective hydrogenation of a-hydroxyketones over cinchona-modified Pt: influence of reactant and modifier structure
O.J. Sonderegger, G.M.-W. Ho, T. Bürgi and A. Baiker
Journal of Molecular Catalysis A: Chemical, 229 (1-2) (2005), p19-24


DOI:10.1016/j.molcata.2004.11.003 | unige:14726 | Article HTML | Article PDF
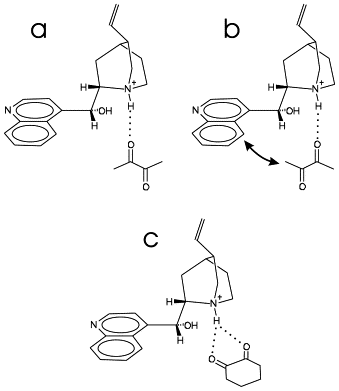
The scope of the asymmetric hydrogenation of functionalized ketones over cinchona-modified platinum was extended to achiral α-hydroxyketones. Cinchonidine showed by far the best catalytic performance affording an enantiomeric excess between 57 and 82% depending on the substrate. O-methoxy-cinchonidine showed poor enantioselection. O-phenoxy-cinchonidine favoured the opposite enantiomer compared to cinchonidine. Solvents with empirical solvent parameters ET N â ranging from 0.10 to 0.65 were tested. Tert-butylmethylether proved to be the most suitable. The highest ratio of substrate/cinchonidine where no loss in e.e. was observed was at around 540, independent of the structure of the α-hydroxyketone. The oxygen in α-position to the ketone seems to play an important role in the enantioselection as well as a phenyl ring or a rigid cis-conformation. The dependence of the enantiomeric excess on the modifier structure and the inversion of the sense of enantiodifferentiation is interpreted in terms of repulsive interactions, which become more evident as the steric demand of the functional group (OH, O-Me, O-Ph) of the modifier increases. The findings indicate that a hydrogen bond in the modifier reactant complex involving the hydroxyl functionality of cinchonidine is not crucial in order to achieve high enantioselectivity.